


연구소 네이처 논문중에 처음으로 Author에 이름을 올린 논문!! 비록 지금은 중간이지만 언젠가는...
Abstract
Massively parallel sequencing technologies have identified a broad spectrum of human genome diversity. Here we deep sequenced and correlated 8 genomes and 7 transcriptomes of unrelated Korean individuals. This has allowed us to construct a genomewide map of common and rare variants and also identify variants formed during DNA-RNA transcription. We identified 9.56
million genomic variants, 23.2% of which appear to be previously unidentified. From transcriptome sequencing, we discovered 4,44 transcripts not previously annotated. Finally, we revealed ,809 sites of transcriptional base modification, where the transcriptional landscape is different from the corresponding genomic sequences, and 580 sites of allele-specific expression. Our findings suggest that a considerable number of unexplored genomic variants still remain to be identified in the human genome, and that the integrated analysis of genome and transcriptome sequencing is powerful for understanding the diversity and functional aspects of human genomic variants.
NG : http://www.nature.com/ng/journal/vaop/ncurrent/full/ng.872.html
'ººº::Learning™:: > ::BioInformatics™::' 카테고리의 다른 글
| [Paper] Reference-unbiased copy number variant analysis using CGH microarrays (0) | 2010.12.02 |
|---|---|
| [Paper] TIARA : a database for accurate analysis of multiple personal genomes based on cross-technology (0) | 2010.12.02 |
| [Analysis] BLAST 프로그램 비교 (0) | 2010.02.04 |
| [Analysis] BLAST 설치 (0) | 2010.01.15 |
| [Article] '아시안 게놈 로드' 프로젝트 (0) | 2010.01.11 |
이 논문은 우리끼리 CARA라 명명한 논문이다.
'ººº::Learning™:: > ::BioInformatics™::' 카테고리의 다른 글
'ººº::Learning™:: > ::BioInformatics™::' 카테고리의 다른 글
| [Paper] Extensive genomic and transcriptional diversity identified through massively parallel DNA and RNA sequencing of eighteen Korean individuals (0) | 2011.07.04 |
|---|---|
| [Paper] Reference-unbiased copy number variant analysis using CGH microarrays (0) | 2010.12.02 |
| [Analysis] BLAST 프로그램 비교 (0) | 2010.02.04 |
| [Analysis] BLAST 설치 (0) | 2010.01.15 |
| [Article] '아시안 게놈 로드' 프로젝트 (0) | 2010.01.11 |
Blast의 5가지 프로그램
- blastp : 단백질 – 단백질 비교 / 관련성이 먼 것을 찾기 위해 치환행렬 사용
- blastn : 염기 – 염기 비교 / 관련성이 멀지 않으며 아주 점수가 높은 매치에 사용
- blastx : 염기(번역) – 단백질 비교 / 새로운 DNA 서열 및 EST 분석에 응용
- tblastn : 단백질 – 염기(번역) 비교 / DB중 코딩 부위를 찾는데 유용함
- tblastx : 염기(번역) – 염기(번역) 비교 / EST분석에 유용하나 많은 계산을 요함
'ººº::Learning™:: > ::BioInformatics™::' 카테고리의 다른 글
대학원 시절 이후로 다시 BLAST를 만지게 될지는 몰랐지만 앞으로 계속 자주 만져야 할거 같아서 처음부터 정리를 해보자 그 첫번째로 설치부터!!
BLAST는 웹사에서 사용할 수 있는 Web BLST와 사용자 컴퓨터 혹은 서버에서 사용 가능한 Local BLAST로 나뉠 수 있다. 그중에서 이번에는 Local BLST설치 및 사용법에 대해 정리해 보자( 사실 Web BLAST는 어찌 설치했는지 기억이 안나서~~ㅋㅋㅋ)
1. BLAST Download & Install
BLAST는 NCBI FTP 사이트에서 다운로드 받을 수 있다. 자신의 컴퓨터 OS 버전에 따라 원하는 BLAST를 다운로드 받는다.(이곳에서는 Linux를 기반으로 설명한다.)
원하는 버전의 BLAST를 다운로드 받았으면 원하는 폴더에 압축을 푼다.
2. Local BLAST를 이용한 Sequence Search
1. 먼저 검색할 서열 데이터베이스가 필요하다. 이는 원하는 서열 데이터베이스를 NCBI혹은 해당 사이트로 부터 다운받자
2. 다운받은 Raw데이터 혹은 Fasta 형식의 데이터를 이용해 BLAST에서 사용가능한 DB Format으로 변경을 하자. 이는 BLAST 검색 속도를 향상기키기 위해 필요한 단계이다.
특히 -p 옵션은 만들어지는 db가 nucleotide냐 protein이냐를 정하는 옵션이다. 이 옵션에 따라 blast program을 실행시킬 수 있다. default는 protein이니 blastn을 사용하기 위해서는 꼭 -p F 로 db를 만들어 주어야 한다.
3. BLAST Search
1. 첫번째로 검색하고자 하는 서열 정보를 Fasta format으로 만들어 준다.
ACGTTTTTTTTTAAAAGAGAGAGAGAGCCCCCCCCC
4. 검색된 결과를 확인한다.
'ººº::Learning™:: > ::BioInformatics™::' 카테고리의 다른 글
조선일보·서울대 유전체의학硏 '아시안 게놈 로드' 프로젝트
아시아 9개국 1000여명 DNA 유전정보 전체 해독
한국인의 눈에는 몽고주름이, 엉덩이에는 몽고반점이 있다. 몽골 사람이나 북미 인디언도 마찬가지다. 한국인과 북방계 유목민족의 연관성을 보여주는 증거다. 반면 고인돌과 솟대는 남방계 아시아인의 흔적이다. 유목민족은 석관묘를 쓴다. 한국인의 뿌리는 북방계인가, 남방계인가.

지난해 7월 서울대 의대 서정선 교수는 30대 한국인 남성의 게놈 전체를 해독한 결과를 '네이처'지에 발표했다. 서 교수는 "한국인은 앞서 게놈이 해독된 남방계 중국인과 뚜렷하게 구분되는 북방계 아시아인임을 확인했다"며 "아프리카인과 유럽인, 남방계 아시아인, 북방계 아시아인이라는 인류를 구성하는 4개 인종의 게놈정보를 완성한 것"이라고 밝혔다.
지난달 11일 한국생명공학연구원을 비롯한 국제컨소시엄이 '사이언스'지에 발표한 연구결과는 그와 정반대였다. 컨소시엄은 "아시아 73개 민족의 개인별 유전자 변이(SNP)를 분석한 결과, 아프리카에서 비롯된 인류의 조상이 인도 북부를 거쳐 동남아시아에 정착했고, 그 중 북쪽으로 이동한 한 갈래가 만주를 거쳐 한반도로 들어왔다"고 밝혔다. 한국인의 기원이 남방계란 말이다.
하지만 두 연구 모두 한계를 갖고 있다. 서 교수팀의 연구는 한국인의 게놈 전체를 해독했다는 점에서 의미가 있지만, 1명만 분석한 것이어서 대표성이 떨어진다. 사이언스지에 실린 연구는 분석대상이 방대하다는 점에서 서 교수팀을 앞서지만, 몽골이나 만주 등 북방계 아시아인은 분석하지 않아 '반쪽짜리 연구'라는 평가를 받고 있다.
조선일보와 서울대 유전체의학연구소가 함께 추진할 '아시안 게놈 로드'는 두 연구의 한계를 모두 극복할 것으로 기대된다. 북방계와 남방계 아시아 국가가 다 들어갔고, 해독 대상도 1000명이나 된다. 사이언스지 논문이 게놈의 극히 일부만 분석했지만, 아시안 게놈 로드는 게놈 전체를 해독할 계획이다. 규모와 정확도에서 명실상부한 최초의 '아시아인 유전자 지도'를 만들 수 있다.
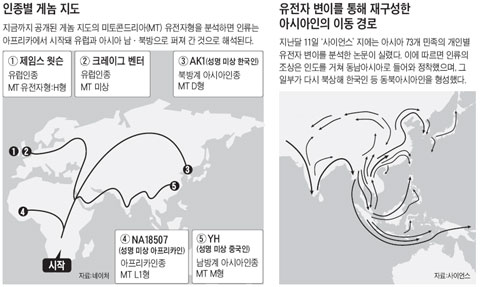
아시안 게놈 로드의 총책임자인 서정선 교수는 "현재로선 한국인의 DNA는 북방계와 남방계의 혼합형으로 추정된다"고 밝혔다. 이는 게놈 일부를 분석한 다른 연구에서 확인된다. 김욱 단국대 생체표지연구센터장은 아시아 각 민족의 미토콘드리아 유전자를 분석했다. 미토콘드리아는 세포핵 밖에 있는 기관으로 난자를 통해서만 후손에게 전달된다. 모계 혈통을 알 수 있다. 세포핵 유전자엔 모계와 부계가 섞여 있다.
미토콘드리아 DNA는 한국인의 60~70%가 북방계 유전자 특성을 보였다. 하지만 세포핵의 개인별 유전자 변이를 분석하면 반대로 60~70%가 남방계였다. 김 센터장은 "한국인은 북방계와 남방계가 혼합돼 형성된 것"이라며 "지난달 사이언스 논문은 약 3만~4만년 전에 남방계 인류가 한반도로 들어왔다는 것이지, 남방계가 한국인의 뿌리라는 의미는 아니다"라고 말했다.
남방계가 오기 전인 약 2만년 전에 이미 북방계가 한반도로 들어왔기 때문이다. 2008년 연세대 신경진 교수팀은 박물관에 있는 구석기·신석기·청동기·백제·신라·고려 시대의 유골 35점에서 미토콘드리아 DNA를 추출해 분석했다. 그 결과 선사시대 한국인의 DNA는 북방계의 특성을 보였으며, 남방계 DNA는 비교적 최근에 유입된 것으로 나타났다. 서정선 교수는 "아시안 게놈 로드는 게놈 전체를 해독해 북방계와 남방계의 혼합 과정을 더욱 세밀하게 보여 줄 것"이라고 말했다.

- ▲ 서울대학교·서정선 교수
◆아시아인용 맞춤 의약 토대 마련
아시안 게놈 로드는 한국인의 기원을 밝혀줄 뿐 아니라 아시아인용 맞춤 의약의
토대도 마련할 것으로 기대된다. 같은 약이라도 아시아인과 서양인에서 효능이 다르게 나타난다는 사실이 잇따라 확인되면서 다국적 제약사들의 아시아
임상시험이 크게 늘고 있다. 한 예로 아스트라제네카가 1996년 개발한 폐암치료제 '이레사'는 서양인 대상 임상시험에서는 별 효과가 없었지만,
아시아인에서는 말기 폐암 환자들에게 효과가 있는 것으로 나왔다. 또 노바티스의 고혈압 치료제 '디오반'은 아시아인 대상 임상시험에서 뇌졸중
예방효과가 입증됐다. 아시안 게놈 로드를 통해 아시아인 고유의 유전적 특성이 밝혀지면, 아시아인에게 맞는 약품이나 치료법이 개발될 수 있다.